Research Area
Abstract
The molecular mechanism of neurodegenerative diseases such as motor neuron disease (amyotrophic lateral sclerosis, ALS) remains unknown. Therefore, therapeutic strategy has not been established. Our laboratory aims to elucidate the mechanism of onset and progression of motor neuron disease, which have been shown to be derived from the pathological changes within different cell types; motor neurons and glial cells. We will analyze inherited ALS, using mouse, cell culture, and in vitro system as models. Based on these studies, we expect to design the therapeutic interventions for the sporadic ALS patients in future.
We have revealed that non-cell-autonomous effect mediated by glial cells around neurons accelerates disease progression in ALS. Therefore, it is important to understand the role of glial cells to develop a novel therapeutic strategy for ALS. We are also focused on the pathological role of neuroinflammation induced by glial cells in Alzheimer's disease.
Non-cell autonomous neurodegeneration
Transgenic mice carrying human SOD1 (superoxide dismutase 1) mutations linked to familial ALS have widely been used for ALS research. In our laboratory, we established LoxSOD1G37R mice, which enable us to eliminate the mutant SOD1 gene from specific cell types, to reveal the cell types involved in ALS onset and progression. Mutant SOD1 in motor neurons determines the ALS onset. On the other hand, mutant SOD1 in glial cells, such as microglia or astrocytes, determines the ALS progression (Boillee et al. Science 2006; Yamanaka et al. Nat Neurosci 2008). These findings indicate that non-cell-autonomous mechanisms induce motor neuron degeneration in ALS (Yamanaka et al. PNAS 2008). We are now striving to elucidate the detailed roles of various cells in the central nervous system on the ALS pathomechanisms.
Research Projects
A) glial cells, neuroinflammation, systematic homeostasis and neurodegenerative diseases
1. Roles of glial cells, neuroinflammation, gila-immune interaction in ALS and AD

Figure. Assumed mechanism of non-cell autonomous motor neuron death in ALS (spinal cord – periferal immune system interactions)
In ALS, not only accumulation of pathological changes inside motor neurons but also pathological changes in glial cells,
such as decreased clearance of glutamate by astrocytes and release of reactive oxygen species or cytotoxic cytokines by microglia,
may further damage motoneurons and exacerbate the pathology. Cell death of oligodendrocytes is also involved in ALS pathogenesis.
Furthermore, recent studies suggest that infiltrating immune cells, which are not originally derived from central nervous system, may regulate microglial cytotoxic activity.
*Bold arrows indicate motoneuron-injurious and dotted arrows indicate motoneuron-protective effects.
Activated glial cells (astrocytes and microglia) in ALS/AD accelerate neurodegeneration by secreting reactive oxygen species (ROS) or proinflammatory cytokines. Recent studies have revealed that infiltrated immune cells, including T cells, regulate microglial activation to protect neurons. These non-neuronal cells are good targets for a novel therapeutic approach for ALS/AD. Using ALS model mice (mutant SOD1 transgenic mice) and next-generation AD model mice (humanized amyloid precursor protein (APP) with familial AD mutations knocked-in mice), we are developing a method to regulate the non-neuronal cells.
2. Exacerbation factor of ALS: TGF-β1
The expression level of TGF-β1 (Transforming Growth Factor β1), which is well known as an anti-inflammatory cytokine, is increased in astrocytes in ALS model mice and human sporadic ALS cases. Overexpression of TGF-β1 in astrocytes shortened survival times of ALS model mice, while an inhibitor of TGF-β1 extended. The overexpression of TGF-β1 reduced the number of infiltrated T cells and neuroprotective activity of microglia, suggesting that TGF-β1 negatively regulates a neuroprotective environment of spinal cords. (Endo et al. Cell Reports, 2015). We are now developing a novel therpeutic strategy targeting TGF-β1.
3. Innate or aquired immune systems in ALS and AD pathogenesis
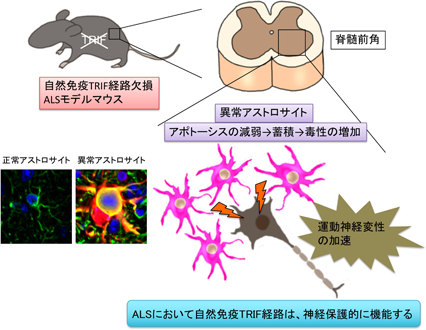
Figure. TRIF pathway, one of the major pathways in innate immune responses, is protective in ALS
Immune responses can be divided into two major categories: innate immune responses and acquired acquired immune responses involving lymphocytes. Until now, the involvement of innate immune responses in ALS pathogenesis has been unclear. To elucidate the role of innate immune responses, we generated ALS mice lacking MyD88 and TRIF, which are key molecules for the function of Toll-like receptors, sensors of innate immune responses. The results showed that only in the absence of TRIF, the survival time of ALS mice was significantly shortened and aberrant astrocytes accumulated in the lesions. This study revealed a novel function of TRIF in the involvement of the innate immune system in ALS and in the removal of aberrantly activated astrocytes at the lesion (Komine et al. Cell Death Differ 2018). We are currently generating ALS and AD model mice with a systemic immune environment shifted to Th1/2 dominance and analyzing how the systemic immune environment alters glial cells and pathology in the brain.
4. Molecular mechanisms of microglial activation in ALS and AD
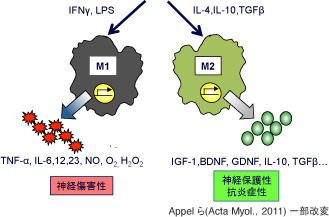
Figure two activated states of microglia, M1 (inflammative) and M2 (anti-inflammative)
Microglia is activated along with motoneuron degeneration. It is thought that two states exist: inflammative M1 with excessive inflammatory cytokines and oxidative stress factors, and neuroprotective M2 with neurotrophic factors and anti-inflammatory cytokines.
It has been suggested that active microglia, which are innate immune cells in the central nervous system, are devided into tow subtypes: neurotoxic (M1 type) and neuroprotective (M2 type), similar to macrophages. In neurodegenerative diseases such as ALS and AD,switching from M2 to M1 may accelerate disease progression (M1/M2 hypothesis). In addition, the concept of disease-associated microglia (DAM) has recently been proposed. It is remain uncovered whether DAMs are neuroprotective or not. It is also uncertain whether DAMs are general in neurodegenerative diseases, remain unresolved. We are searching for factors that induce active microglial conversion based on DAM and M1/2 hypotheses.
B) Intraneuronal mechanisms of motor neuron degeneration
1. Molecular mechanisms of TDP-43 induced neurodegeneration
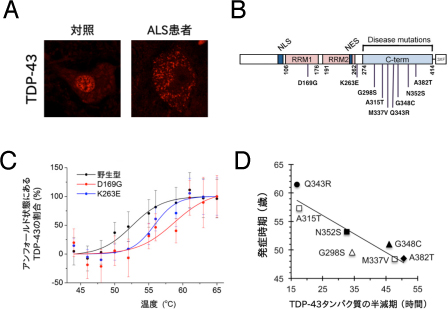
Figure. TDP-43 abnormalities related to ALS pathogenesis
TDP-43 is an RNA-binding protein that regulates splicing, transcription and translation, and is normally localized to the nucleus. However, in sporadic ALS or frontotemporal lobar degeneration (FTLD), TDP-43 leaks into the cytoplasm (Figure A) and forms intracellular inclusions. Some ALS cases are known to be caused by mutations in TDP-43. Most of the mutations identified so far are located on the C-terminal side (Figure B). We have shown that mutant TDP-43 is abnormally stabilized in the cell by enhancing thermal stability (Figure C) and acquiring anti-aggregation properties. Furthermore, we found a negative correlation between the longer half-life of mutant TDP-43 and the earlier onset of disease (Figure D).
TDP-43, an RNA-binding protein, was identified as an abnormally accumulated protein in sporadic ALS (non-hereditary ALS), which accounts for the majority of ALS cases, and frontotemporal lobar degeneration (FTLD), a type of dementia, in 2006. Clarifying the function of TDP-43 and how its abnormalities modulate the motor neurons is the key to understanding the pathogenesis of ALS. We have previously shown that mutant TDP-43 is abnormally stabilized in cells compared to the wild type (Watanabe et al. J Biol Chem 2013) (Austin et al. PNAS 2014). TDP-43 accumulates in Gem, the site of nuclear spliceosome factor maturation, and binds to SMN, the causative gene product of spinal muscular atrophy (SMA), and spliceosome component proteins (snRNPs) aggregate abnormally in sporadic ALS (Tsuiji et al. EMBO Mol Med 2013). We also found in mice that TDP-43 accumulation causes hippocampal inhibitory nerve damage and cognitive decline (Tsuiji et al. Sci Rep 2017). We are currently conducting experiments using cultured cells and mice to elucidate the mechanisms that cause these TDP-43 abnormalities.
2. disruption of organelle-contacting sites in neurodegenrative diseases
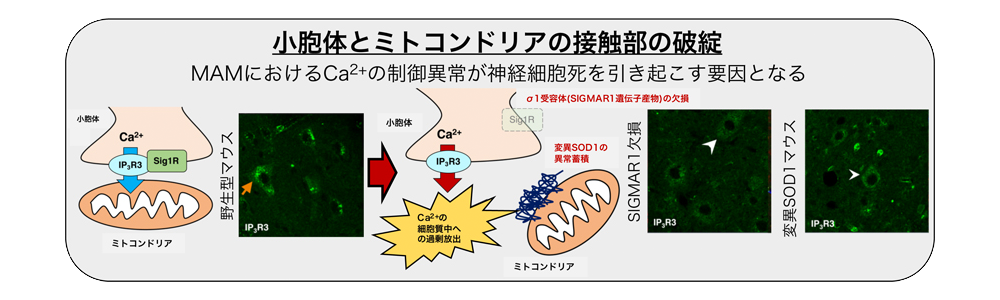
Figure. MAM disruption is related to ALS pathogenesis
Inositol triphosphate receptor type 3 (IP3R3), which transports Ca2+ ions from the endoplasmic reticulum to mitochondria, is normally localized selectively in MAM (orange arrows). (orange arrowheads in the figure), but in the mouse model of ALS pathology, the entire cell is mislocalized (white arrowheads in the figure). This abnormality causes excessive Ca2+ influx into the cytoplasm and Ca2+ deficiency in the mitochondria, resulting in neuronal damage.
Motor neurons require a large amount of energy to perform their functions due to their long axons and large cell bodies, and mitochondria, organelles involved in energy production, are considered to be important. Although organelle abnormalities such as mitochondrial injury and endoplasmic reticulum stress have been proposed as hypotheses for the pathogenesis of ALS, recently, more evidence is accumulating that these organelles are interconnected. In our laboratory, in two different familial ALS models, SOD1 and SIGMAR1, the contact area between the endoplasmic reticulum, which is important for protein synthesis, and mitochondria, which is important for energy production, has been shown to be a key component of the The disruption of the endoplasmic reticulum-mitochondrial intermembrane region (MAM) was found to be a common pathogenesis (Watanabe et al. EMBO Mol Med 2016). Disruption of MAM causes neuronal cell death via dysregulation of intracellular Ca2+, and protecting MAM is expected to be a potential treatment for ALS. We are currently conducting further research to determine whether MAM abnormalities are a universal phenomenon in ALS and to elucidate the molecular mechanisms that maintain and disrupt MAM.