日本語 English
研究概要 Our Reserch Summary
多くの遺伝性神経変性疾患の病因遺伝子が同定されていますが、その発症や疾患進行機序に関してはいまだ解明されていません. 当研究室では神経変性疾患の病態解明を目指し、特に筋萎縮性側索硬化症(ALS)とアルツハイマー病(AD)について研究を行っています. 疾患関連遺伝子を導入したモデルマウス, 培養細胞, in vitro の系を樹立して解析し、その成果を孤発性(非遺伝性)の病態解明に応用します.
当研究室は、これまでALSにおける神経変性の進行を神経周囲のグリア細胞の異常が加速させることを明らかにしました. 神経細胞でおこる病的変化、たとえばタンパク質やRNAの代謝異常, オルガネラ異常などに起因する変化を解明しつつ、周囲のグリア細胞で起こる病的変化を解明することが疾患の進行を制御する、すなわち神経変性疾患の治療戦略を構築するうえで重要であり、これらの解明を目指します. さらに,AD病態におけるグリア細胞が引き起こす神経炎症の役割についても研究をすすめています.
ポイント
非細胞自律性の神経細胞死
Non-cell Autonomous Neurodegeneration
遺伝性ALSの原因遺伝子 SOD1 の遺伝子変異を全身に発現する変異SOD1トランスジェニックマウスは、これまでALSの病態研究に広く用いられてきました. 当研究室では、変異SOD1遺伝子を特定の細胞群から選択的に除去できる独自のモデルマウス LoxSOD1G37R を樹立して、ALSの発症と進行に関与する細胞群を明らかにしてきました. 運動ニューロンにおける変異SOD1がもたらす病的変化は本疾患の発症時期を規定し、グリア細胞であるミクログリア, アストロサイトにおける病的変化はその進行速度を加速することを明らかにしました(Boillee et al. Science 2006; Yamanaka et al. Nat Neurosci 2008). そして、ALSの運動神経変性は神経細胞のみの異常では決まらず、周囲のグリア細胞が主体的に関与して変性の進行が規定される、非細胞自律性(Non-cell autonomous)と呼ばれる性質を示すことを証明しました(Yamanaka et al. PNAS 2008). これらの知見に基づいて、研究室では神経変性疾患の病態を細胞群ごとの視点で解明することを目標としております.
研究プロジェクト Our Reserch Projects
A) グリア細胞・免疫・全身環境と神経変性疾患
1. ALS・ADにおける神経炎症およびグリア-免疫連関の意義
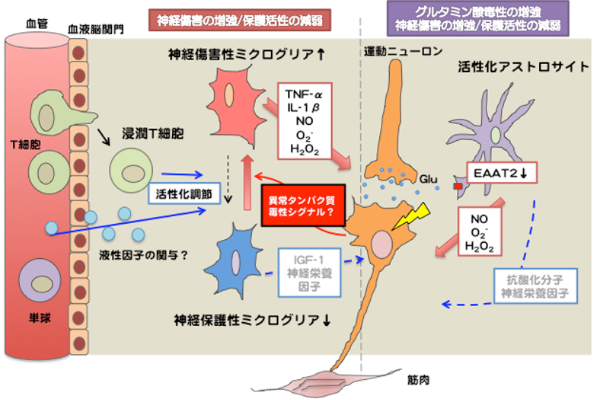
ALSやADの病巣で活性化が見られるグリア細胞(ミクログリア・アストロサイト)は、炎症性サイトカインや活性酸素などの放出を介して神経細胞の変性を加速していると考えられます. また、最近では浸潤するT細胞などの免疫細胞がグリア細胞の活性を制御することで、神経保護的に働くことが示唆されています. これらの非神経細胞を制御することで、疾患の進行を遅らせることができる可能性について研究を進めています. ALSは主として変異SOD1トランスジェニックマウス、ADは次世代ADモデルマウス(ヒト化アミロイド前駆タンパク質(Amyloid Precursor Protein; APP)遺伝子に家族性AD患者由来の変異をノックインしたマウス)を用いて解析しています. また、共同研究で疾患脳や脊髄試料を用いた遺伝子発現解析も行い、モデルマウスの解析に活用します.
2. 自然免疫・獲得免疫系によるALS・AD病態の制御
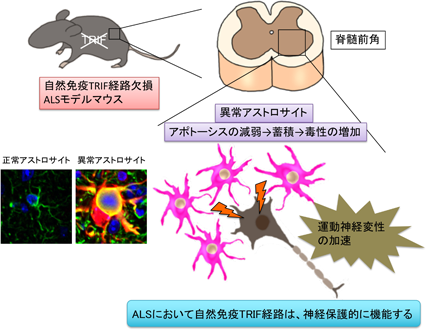
免疫反応は、先天性の自然免疫反応とリンパ球が関与する後天性の獲得免疫反応の2つに大別されます. これまで、自然免疫反応のALS病態への関与は明らかではありませんでした. 当研究室では自然免疫反応の役割を明らかにするため、自然免疫反応のセンサーであるToll様受容体の機能の鍵となる分子MyD88およびTRIFを欠損したALSマウスを作成して解析しました. その結果、TRIFを欠損した場合にのみALSマウスの生存期間が著しく短縮し、異常化したアストロサイトが病巣に蓄積しました. 本研究により、ALSにおける自然免疫系の関与と病巣で異常に活性化したアストロサイトの除去に関わるTRIFの新たな機能を明らかにしました(Komine et al. Cell Death Differ 2018). 現在、全身の免疫環境をTh1/2優位にシフトさせたALSおよびADモデルマウスを作成し、全身の免疫環境が脳のグリア細胞や病態をどのように変化させるかを解析しています.
3. ミクログリアの活性型転換誘導因子の探索
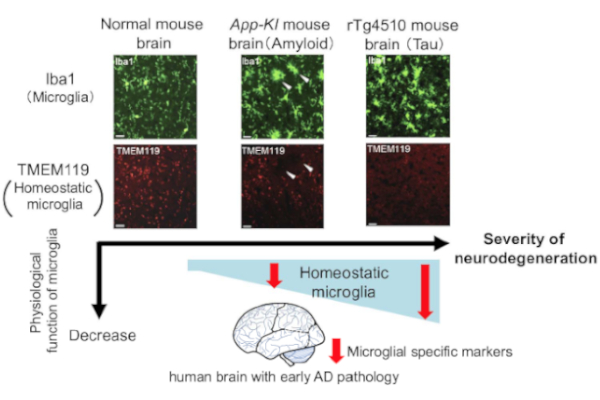
中枢神経系における自然免疫細胞であるミクログリアの活性型には、マクロファージと同様、神経傷害性のM1型および神経保護性のM2型の存在が示唆されており、神経変性疾患であるALSやアルツハイマー病などにおいてこれらの活性型の転換(神経保護性、いわゆるM2型、から神経傷害性、いわゆるM1型、へ)が疾患の進行を加速する可能性が考えられています(M1/M2仮説). さらに、最近では疾患ミクログリアに共通する活性化(Disease-associated microglia: DAM)という概念が提唱されています. DAMは神経保護性や傷害性をもつのか、本当に神経変性疾患に共通しているのか未解明な点が多いと考えています. 私達は神経変性疾患の病態の進行を遅延させる分子群を同定するため、DAMやM1/2仮説を手がかりにミクログリアの活性型転換誘導因子を探索する研究を行っています.
B) 神経細胞内環境の破綻と神経変性機序の解明
1. TDP-43の異常がALSを引き起こすメカニズムの解明
ALSの大部分を占める孤発性ALS(非遺伝性のALS)や認知症の一種である前頭側頭葉変性症(FTLD)の病巣に蓄積するRNA結合タンパク質TDP-43が2006年に同定されました。 TDP-43の機能やその異常が運動神経にどのような変調をもたらすのかを明らかにすることは、ALSの病態解明の本丸であると考え、当研究室でもその解析をすすめています. これまでに変異TDP-43は野生型よりも細胞内で異常に安定化すること(Watanabe et al. J Biol Chem 2013)やTDP-43が核内スプライソソーム因子の成熟の場であるGemを破綻させること(Tsuiji et al. EMBO Mol Med 2013)を見出しました. 現在、これらTDP-43の異常を引き起こすメカニズムの解明を目指し、培養細胞やマウスを用いた実験を行っています.
ポイント
TDP-43単量体化
TDP-43 Monomerization
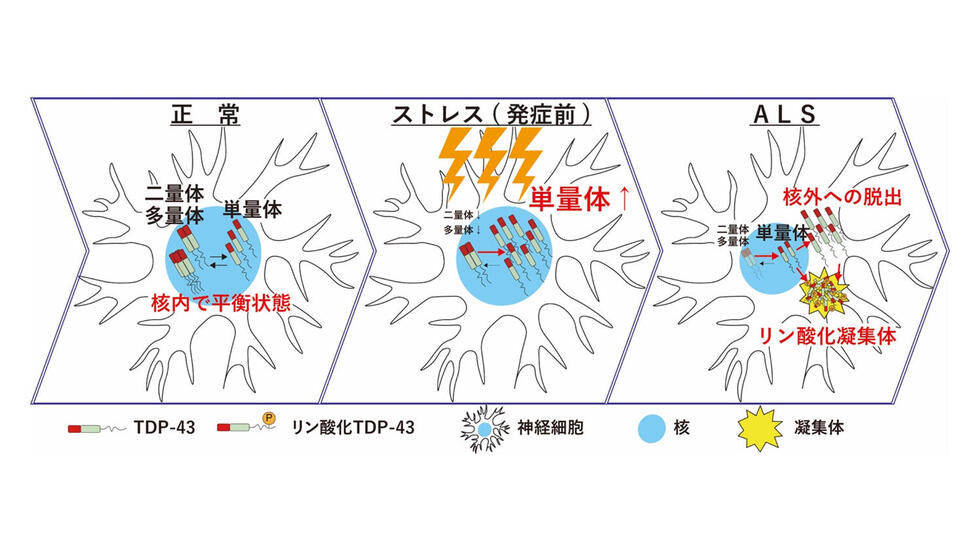
我々は最近、TDP-43の単量体化がALSにおけるTDP-43異常化の引き金となることを世界に先駆けて発見しました(Oiwa et al. Sci Adv 2023). TDP-43は、通常、核内でTDP-43同士が結合したホモ多量体として存在し、遺伝子の発現制御などの重要な機能を担っています. しかし、神経細胞に様々なストレスがかかることでTDP-43の多量体化が阻害されると、TDP-43は核から細胞質へと流出し、ALS患者で見られるような異常な凝集体を形成することが判明しました. TDP-43の単量体化を抑制できればALSにおける新たな治療法開発が可能になり、さらにTDP-43の単量体化を検出することでALSの早期診断と早期治療が可能になると期待されます.
2. 神経変性におけるオルガネラ連関破綻の意義の解明
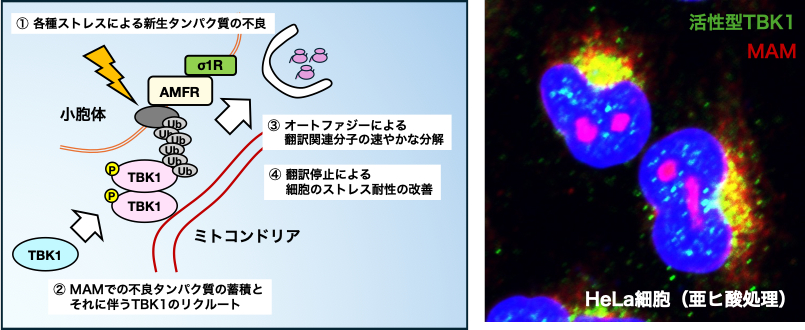
運動神経細胞は軸索の長さと細胞体の大きさという特徴から、その機能発揮に多大なエネルギーを必要としており、エネルギー産生に関わる細胞内小器官(オルガネラ)であるミトコンドリアは重要であると考えられています. 最近ではこれらのオルガネラがお互いに相互作用し、その相互作用(連関)の破綻が疾患を引き起こしているという知見が蓄積しています. 当研究室では、タンパク質合成に重要な小胞体とエネルギー産生に重要なミトコンドリアの接触部分「小胞体・ミトコンドリア膜間領域(MAM)」の破綻に着目し、ALS病態におけるMAM破綻の重要性を明らかにしてきました(Watanabe et al. EMBO Mol Med 2016, Watanabe & Horiuchi et al. Neurobiol Dis 2023 ほか). MAMの破綻は細胞内Ca2+の制御異常を介して神経細胞死を引き起こしており、MAMを保護することでALSの治療が可能になると期待されます. 最近では、MAMの破綻がALS原因遺伝子産物であるTBK1の機能を阻害し、異常化したタンパク質に対して細胞を脆弱化させることを報告しました(Watanabe et al. PNAS 2023). 現在、MAMを維持・破綻させる分子メカニズムの解明を目指し、更なる研究を行っています.
このほかにも様々な視点からALS・ADの病態解明と治療法開発に向けた研究を進めています。研究内容に興味を持たれた方は、随時、下記メールアドレスまで御連絡下さい。